GOeval
GOeval.RmdFirst, install and load this package. It is easiest to use either the “devtools” or “remotes” package to do so. “remotes” is a smaller package, so it might be the better option if you do not want to install all of “devtools.”
devtools::install_github("BrentLab/GOeval")
# OR
remotes::install_github("BrentLab/GOeval")Now that the package is loaded, we need a network as a tab-separated .tsv file where the first column is the source nodes, the second column is the target nodes, and the optional third column is the edge scores.
To run the following examples, access the example data stored in “vignette_data.tar.gz” and extract it into a local folder.
data_path <- system.file(package = "GOeval", file.path("vignette_data.tar.gz"))
# wherever you want to store the example data
data_dir <- "GOeval_data"
untar(data_path, exdir = data_dir)Basic evaluation:
Here are the required arguments to run the evaluate function on the “small_np3.tsv” network, which includes 10 transcription factors.
# path to a network in proper format
network_path <- file.path(data_dir, "small_np3.tsv")
# path to a .txt file with one column that includes all the genes
# that may appear in the network
reference_path <- file.path(data_dir, "h1_k562_overlap_universe.txt")
# wherever you want to store the generated output
out_dir <- "GOeval_output"
# a short name for the network without any spaces
network_name <- "np3"Run the evaluate function.
net_1_data <- evaluate(network_path, reference_path, out_dir, network_name,
# some additional arguments
edges = c(512, 1024, 2048, 3174, 4096, 8192, 16384, 32768, 65536),
permutations = 1,
get_annotation_overlap = TRUE
)It is possible to customize the behavior of the evaluate
function by using arguments in addition to the four required ones. In
the above example, the ‘edges’ argument modifies the sizes of subsets to
analyze from the input network. “permutations = 1” allows a faster run
time than the default “permutations = 3” at the cost of running the
analyses on fewer permuted versions of the input network.
“get_annotation_overlap = TRUE” indicates that the “annotation_overlap”
metric should be calculated.
The available arguments for all functions can be found at the Reference tab at the top of this page.
After evaluate runs for a couple minutes, the directory
at out_dir should contain a folder of subsets of the original network, a
folder that contains the ORA summaries and sets of target genes, and a
.pdf file with plots of metrics calculated from those summaries. The
evaluate function returns the data used to make the plots as a list of
data.frames.
The run time of the evaluate function changes with the size of the
network and the number of permutations. With larger networks, it may
take up to several hours. It is thus recommended to use a computing
cluster when working with large networks. It is possible to speed up the
process if performing each step manually as shown in the “Advanced
usage” section below and running the webgestalt_network
function calls in parallel.
Now, let us compare with a second network. The “small_dto.tsv” network does not have edge scores, so no network subsets will be created. Generate the data for this network in a similar way as for the first one.
net_2_data <- evaluate(file.path(data_dir, "small_dto.tsv"),
file.path(data_dir, "h1_k562_overlap_universe.txt"),
out_dir,
"dto",
permutations = 1, get_annotation_overlap = TRUE,
# without multiple subsets, the plot would just be one dot
# so we can tell it not to bother
plot = FALSE
)Use the output from both networks in the call to
plot_metrics.
# set main plot titles
title_text <- "NP3 vs DTO"
subtitle_text <- "subset for overlapping TFs"
# perTF controls the x-axis label
# true only if the network subset sizes were determined by
# average number of edges per TF instead of by total number of edges
perTF <- FALSE
plot_data <- plot_metrics(c(net_1_data, net_2_data), title_text, subtitle_text, perTF, annotation_overlap = TRUE)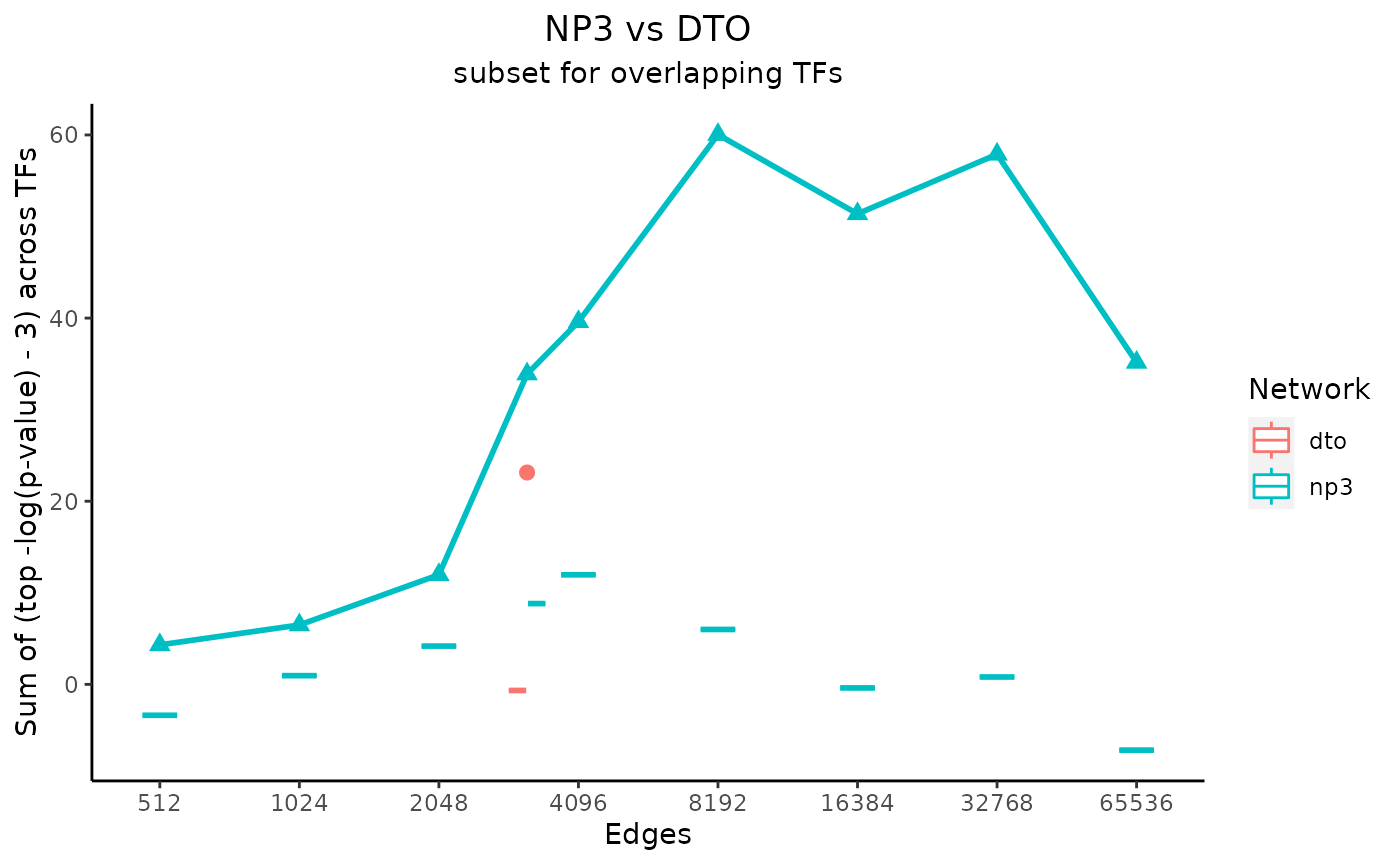
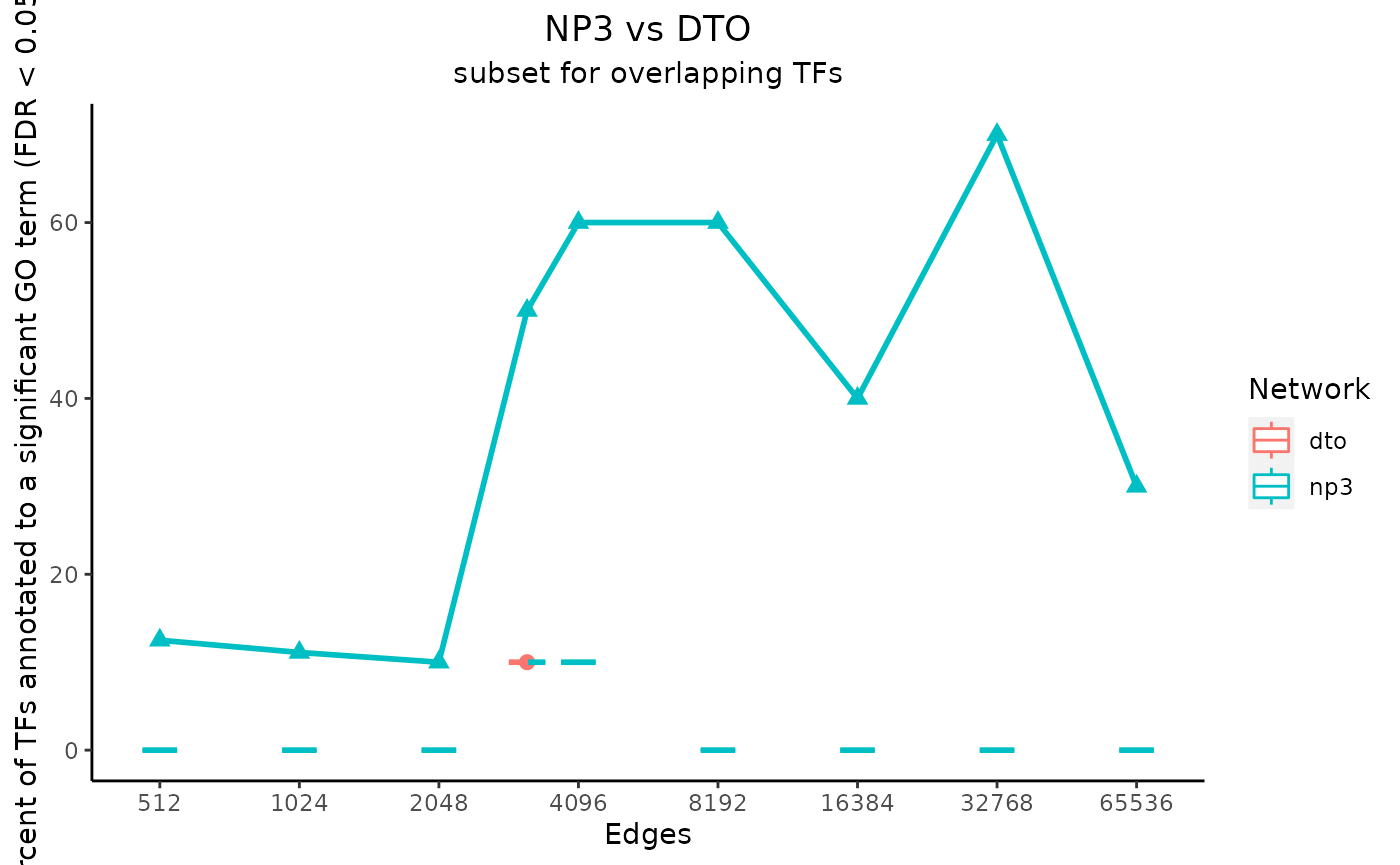
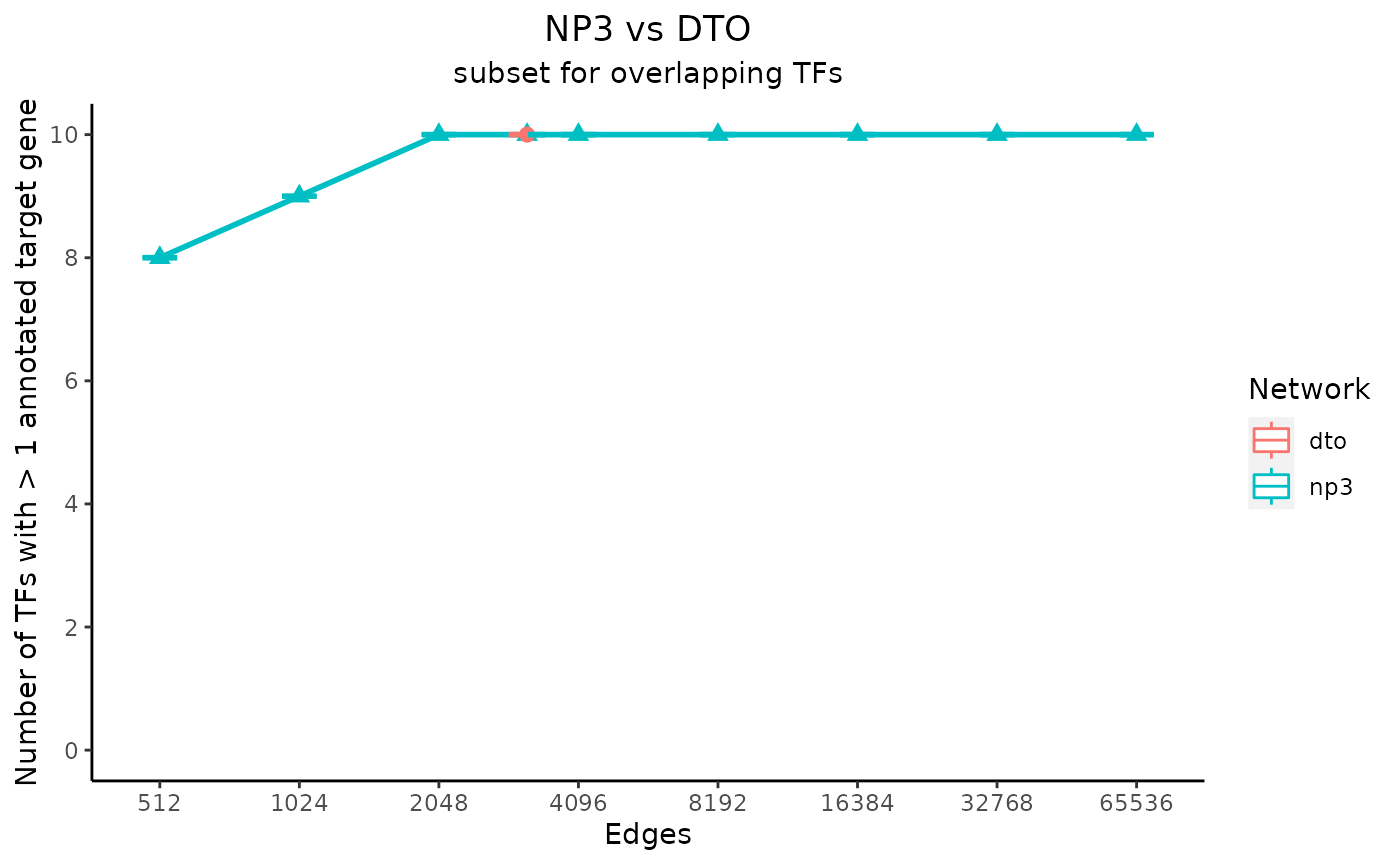
Notice that the metrics for the “NP3” network are plotted as a line, and the metrics for the “DTO” metric are plotted as a dot at one value on the x-axis. This is because the input “NP3” network included a column of edge scores, so multiple subsets were created and analyzed.
In the case that one of the input networks has only one size, the values for the permuted networks are shown as boxplots. They are visible in these example plots as horizontal lines, as only one permuted network was made for each network size. If every input network had multiple subsets, however, the medians of the values for the permuted networks would be plotted as a line for each input network.
Notes on default behavior of evaluate:
By default, only the “sum” and “size” metrics are calculated by
evaluate. Other available metrics are “percent”, “mean”,
“median”, and “annotation_overlap”. Descriptions of each are available
in the function documentation.
For this function to work properly, the correct ‘organism’ and ‘gene_id’ must be specified in accordance with the genes in the input network and reference set. The default values of each are “hsapiens” (human) and “ensembl_gene_id” (for human, “ENSG” followed by 11 numbers). You can see more options for ‘organism’ and ‘gene_id’ with WebGestaltR::listOrganism() and WebGestaltR::listIdType() respectively. The exact gene ID format specified by each option given by WebGestaltR::listIdType() may not be clear. To see examples, navigate to webgestalt.org, select an “Organism of Interest”, and click on the dropdown for “Gene ID Type”. Hovering the cursor over an option for a few seconds will display some example gene IDs of that type. While not ideal, this may be how you have to identify which option for ‘gene_id’ corresponds with the format of the gene IDs in your input.
The ‘database’ argument specifies which database contains the gene set terms in which to search for over-representation. The default is “geneontology_Biological_Process_noRedundant”. See more options with WebGestaltR::listGeneSet(). NOTE: the “annotation_overlap” metric is only designed to work with “geneontology_Biological_Process_noRedundant” or “geneontology_Biological_Process”.
Advanced usage:
All intermediary steps in the evaluation can be run separately with the following functions.
subset_networkto create network subsets with the top-weighted edgeswebgestalt_networkon each of the network subsets to generate ORA resultsget_metricson the stored ORA results to calculate summary statistics for plottingplot_metricsusing the output of get_metrics
We will now walk through step-by-step evaluation of the network “np3_dtoTFs.tsv”, which has 56 TFs and a total of 629,832 edges.
1. subset_network
“np3_dtoTFs.tsv” is tab-separated and contains 3 columns: the TFs, the regulated genes, and the scores.
This function sorts the input network in descending order by the values in the third column and creates files in the output_folder with only the first two columns of the rows that have the highest scores.
input_file <- file.path(data_dir, "np3_dtoTFs.tsv")
output_directory <- file.path(out_dir, "np3_dtoTFs_subsets")
name <- "np3"
# Since we are setting num_possible_TFs, this will be the average number of
# edges from each TF in each subset.
edges <- c(8, 16, 32, 64, 128, 256, 512, 1024, 2048, 4096)
# The "np3_dtoTFs.tsv" network was made to include only 56 TFs.
num_possible_TFs <- 56
subset_network(input_file, output_directory, name, edges, num_possible_TFs)The result is that now the “data/np3_dtoTFs_subsets” folder contains 10 files: “np3_8.tsv” through “np3_4096.tsv”.
webgestalt_network
Run ORA using the webgestalt_network function on each of
the network subsets created in step 1. These files are tab-separated
with the first column containing the TF names and the second column
containing the regulated gene names. “h1_k562_overlap_universe.txt”
contains one column of the names of all genes that could appear in the
network. The network_name should be different for each subset, so I
choose to use the subset file names without the extension.
Note organism = “hsapiens” and database = “geneontology_Biological_Process_noRedundant” is the default database for ORA.
You can see more options for organism and database with WebGestaltR::listOrganism() and WebGestaltR::listGeneSet() respectively.
Also note that the run time of the below code could take well over an
hour, so this vignette does not actually evaluate the code. The run time
is highly dependent on the number of subsets, the number of TFs in each
of those subsets, and the number of permutations. The process can be
sped up by running webgestalt_network on each subset at the
same time using a computing cluster or similar.
for (subset in list.files(file.path(data_dir, "np3_dtoTFs_subsets"), full.names = TRUE)) {
webgestalt_network(
network_path = subset,
reference_set = file.path(data_dir, "h1_k562_overlap_universe.txt"),
output_directory = file.path(out_dir, "np3_dtoTFs_summaries"),
# this just gets the name of each file minus the extension
network_name = strsplit(basename(subset), "[.]")[[1]][1],
permutations = 3
)
}To test the very basics on a personal computer, the following code should run in just a few minutes.
webgestalt_network(
network_path = file.path(data_dir, "np3_dtoTFs_subsets", "np3_8.tsv"),
reference_set = file.path(data_dir, "h1_k562_overlap_universe.txt"),
output_directory = file.path(out_dir, "np3_dtoTFs_single_permutation"),
network_name = "np3_8",
permutations = 1
)get_metrics
Run get_metrics to calculate the desired metrics based
on the output from webgestalt_network. Note the arguments
‘get_sum’ and ‘get_size’ are TRUE by default.
# This would normally be in the 'out_dir' directory, but since this data was
# premade due to computation time constraints, it is available in 'data_dir'
output_folders <- file.path(data_dir, "np3_dtoTFs_summaries")
metric_dfs <- get_metrics(output_folders, get_percent = TRUE, get_mean = TRUE, get_median = TRUE, get_annotation_overlap = TRUE, parallel = FALSE)If you want to compare multiple networks, you can run
get_metrics on both at the same time as shown below with
the addition of the “EDN” network. When comparing multiple networks, the
sizes for all must be based either on total edges or on edges per TF,
but the exact subset sizes do not need to be the same.
# Specify which folders contain the data for each network.
# Each element will be a folder used as the output folder for a call to webgestalt_network.
# Again, these are normally in 'out_dir' but here they are in 'data_dir' for convenience
output_folders <- c(file.path(data_dir, "np3_dtoTFs_summaries"), file.path(data_dir, "EDN_summaries"))
metric_dfs_by_net <- mapply(get_metrics, output_folders, MoreArgs = list(get_percent = TRUE, get_mean = TRUE, get_median = TRUE, get_annotation_overlap = TRUE, parallel = FALSE), SIMPLIFY = FALSE)plot_metrics
Set a title and subtitle for the plots.
Specify whether the subset sizes are based on total edges or edges per
TF.
Set the metric booleans to the same values as those used in
get_metrics to ensure proper plot labels.plot_metrics also returns a list of the data.frames used to
plot. Each data.frame contains values for one metric across all
networks, subsets, and permutations.
# Specify the main titles of the output graphs
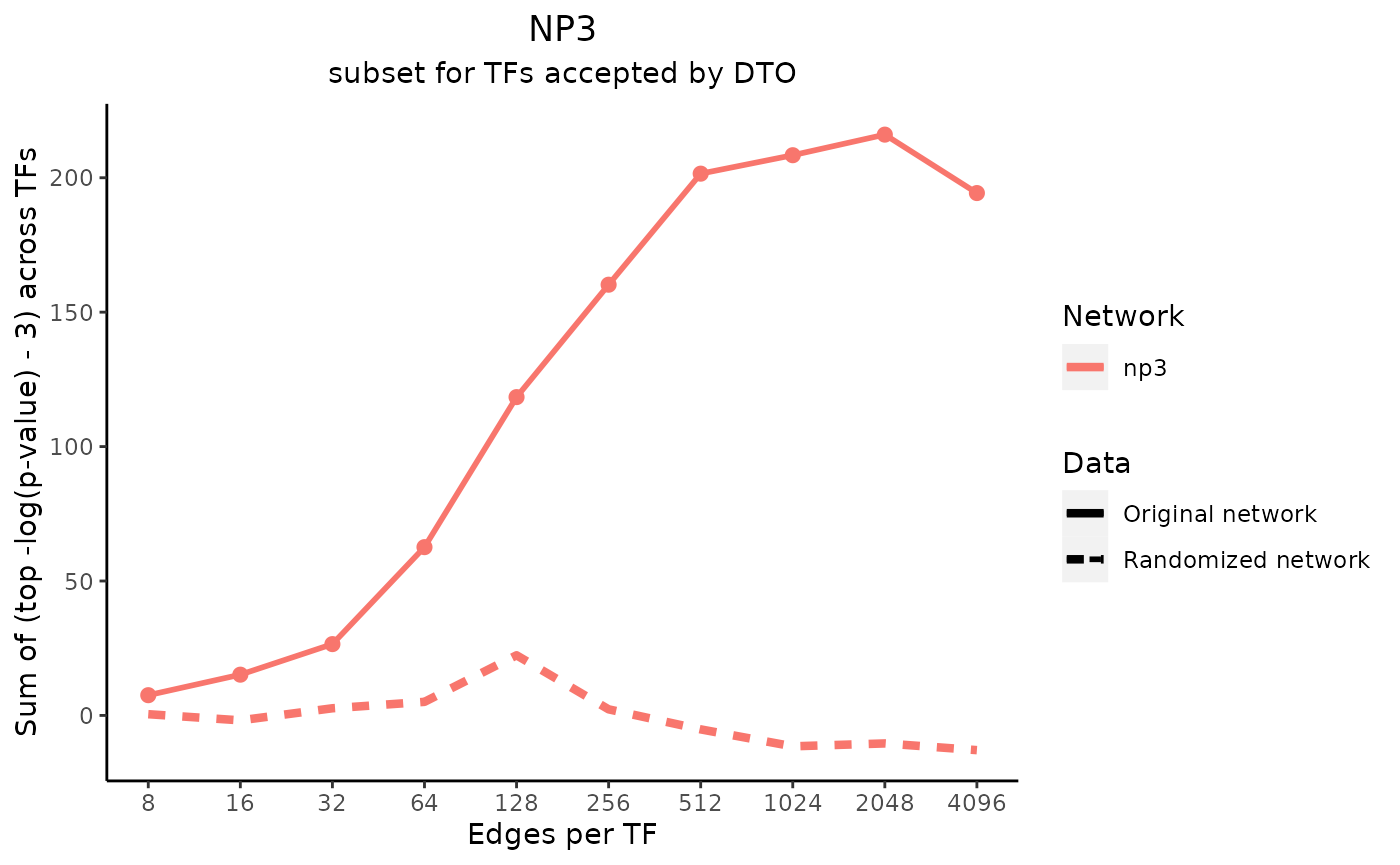
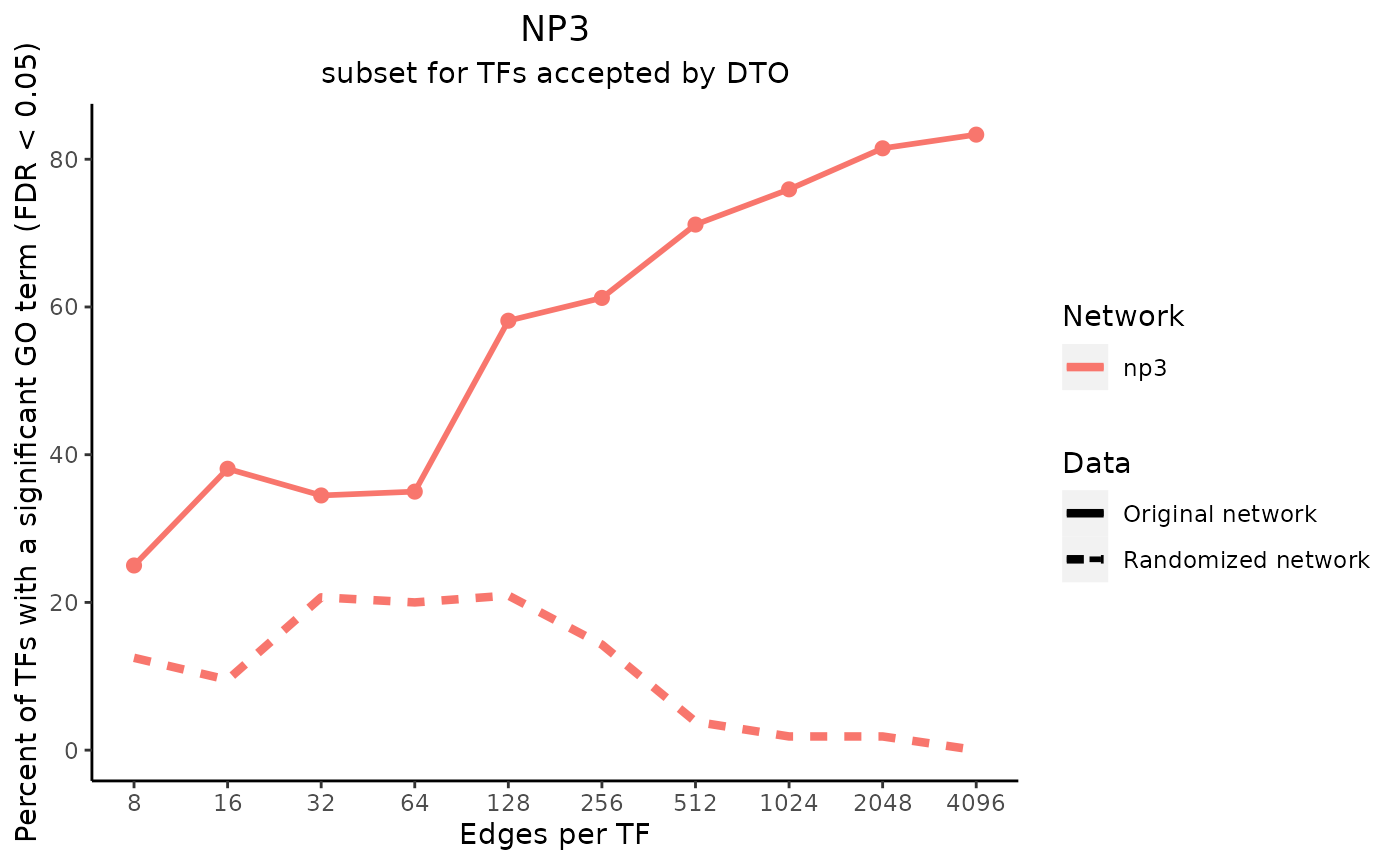
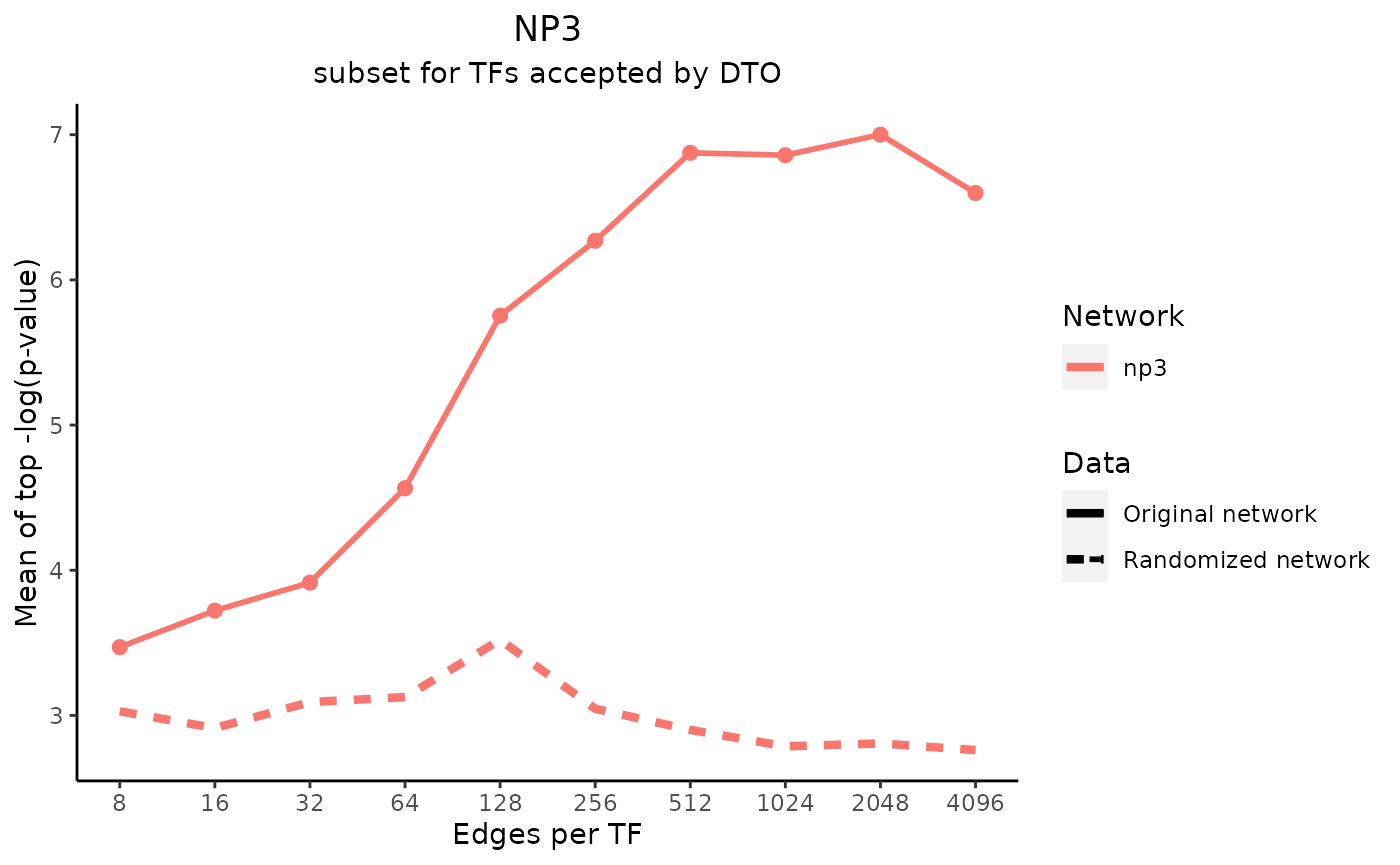
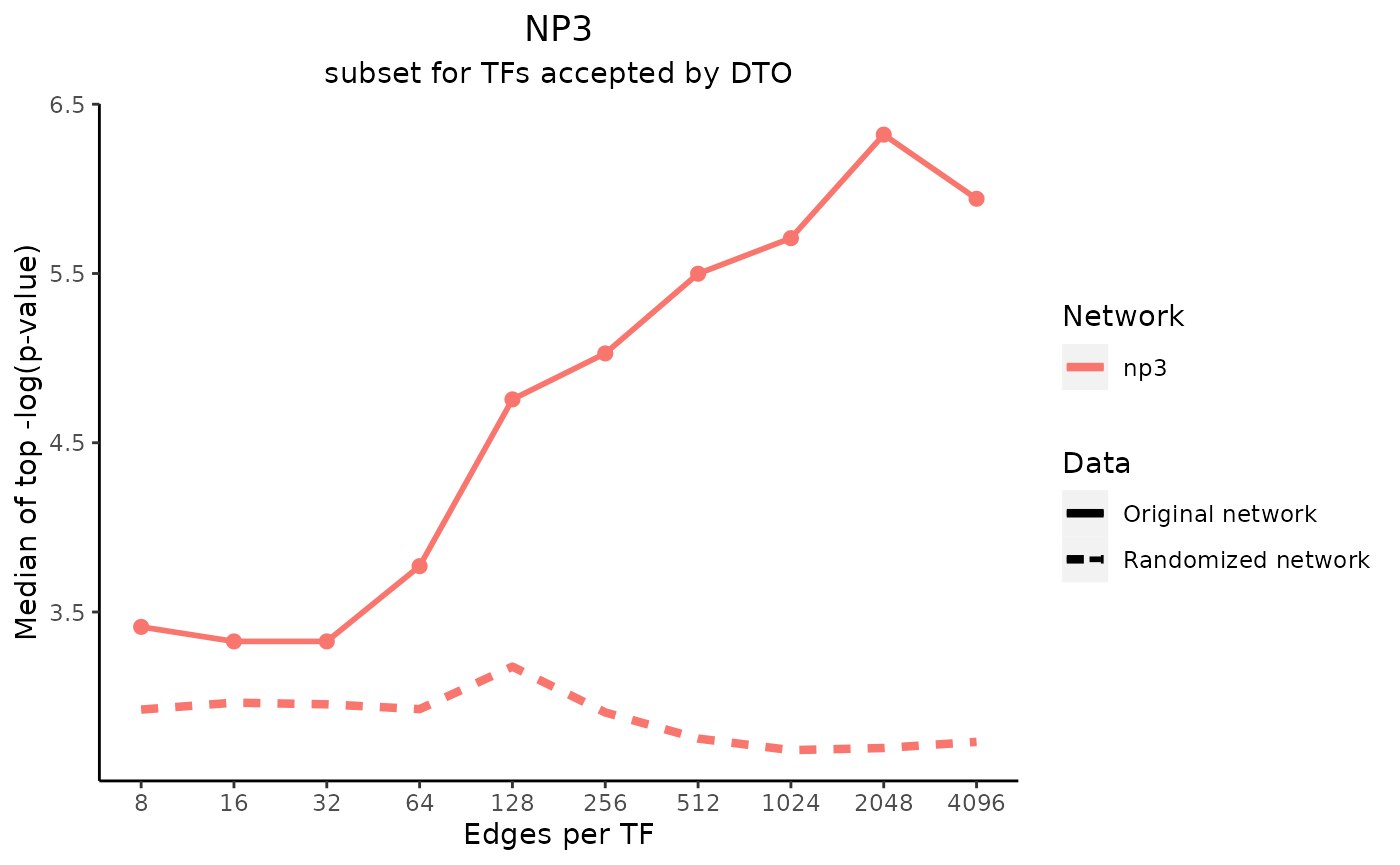
title_text <- "NP3"
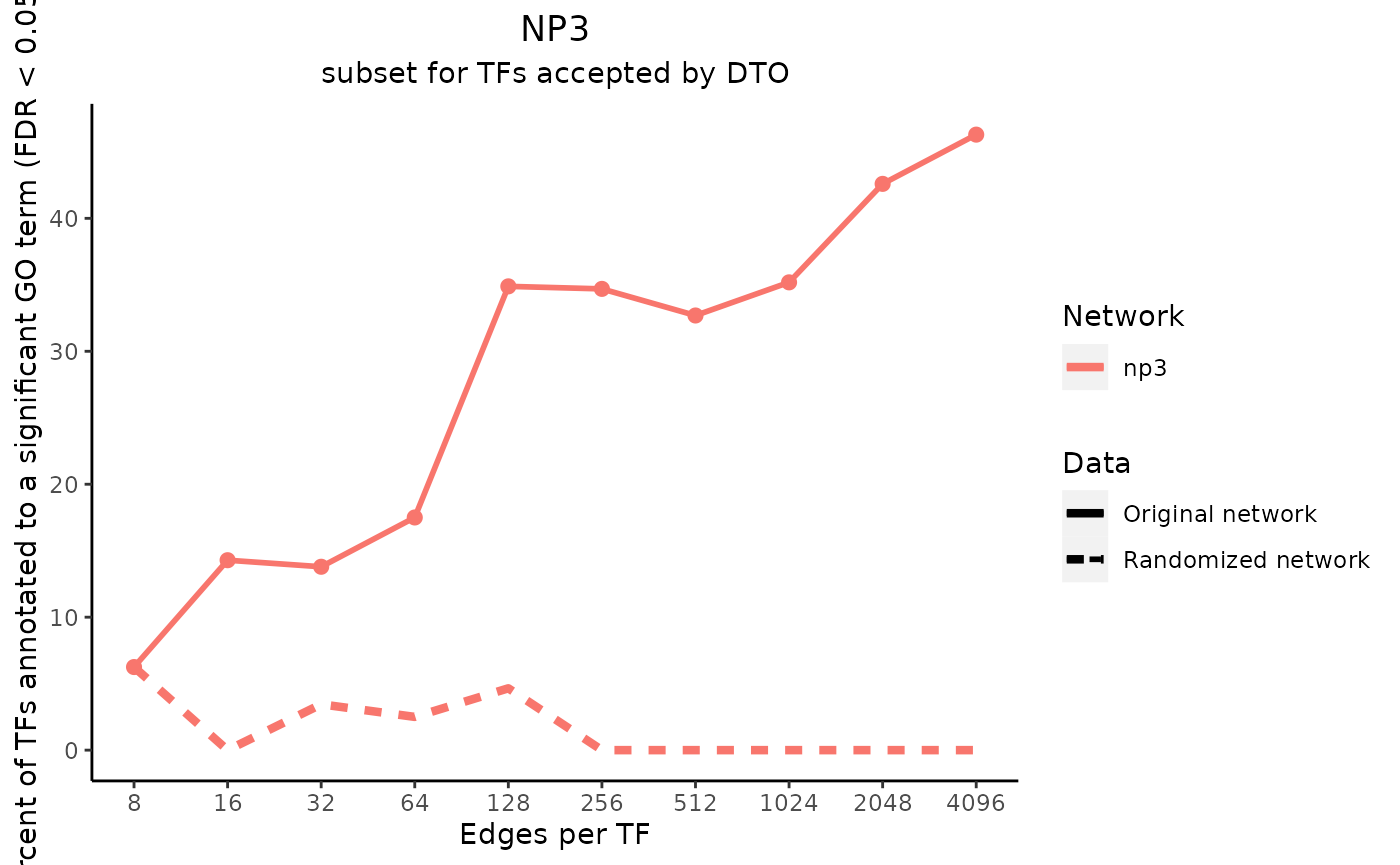
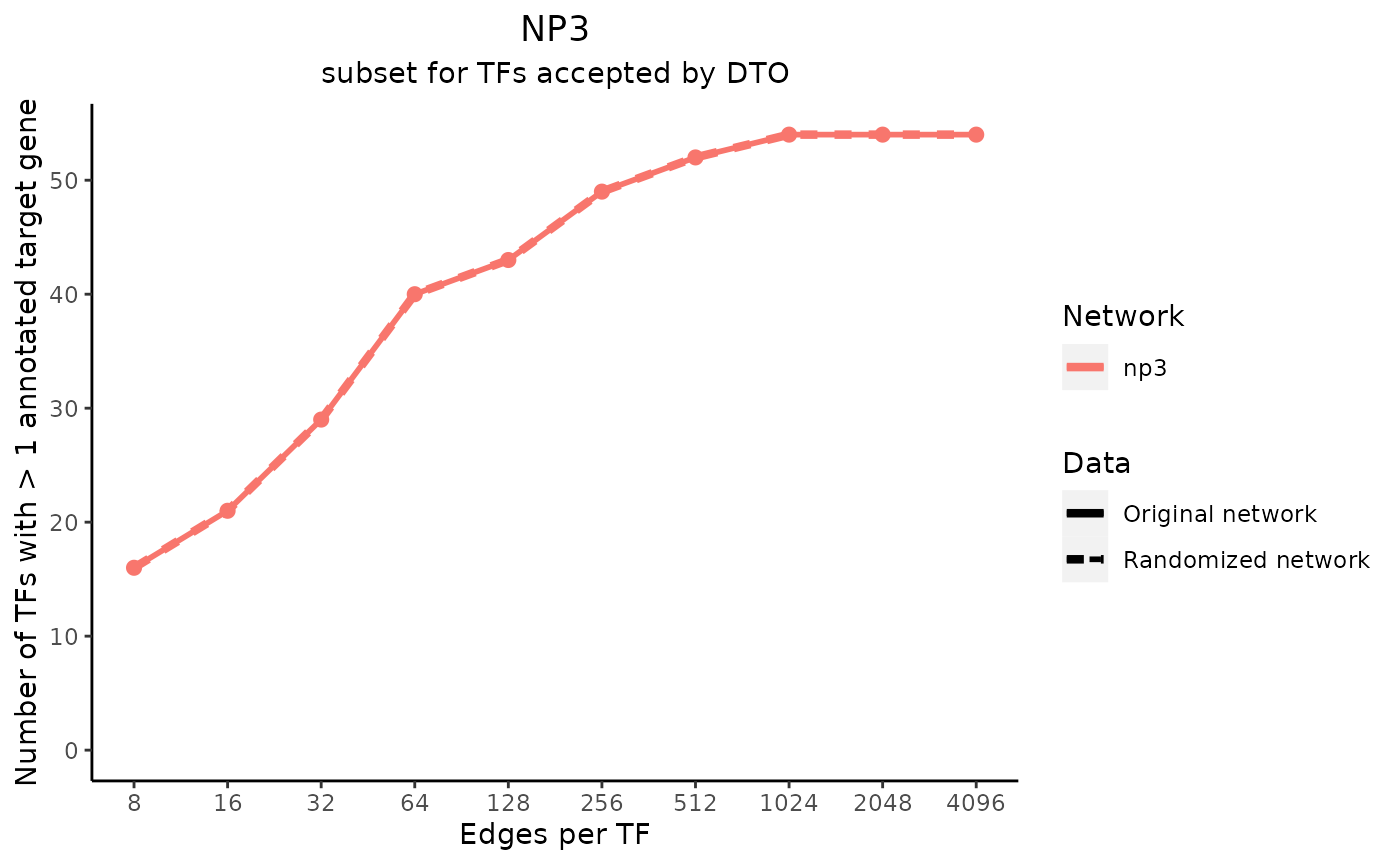
subtitle_text <- "subset for TFs accepted by DTO"
perTF <- TRUE
plot_data <- plot_metrics(metric_dfs, title_text, subtitle_text, perTF, percent = TRUE, mean = TRUE, median = TRUE, annotation_overlap = TRUE)